There are two main pathways to apoptosis:
- Exogenous pathway – activation of the intracellular apoptotic enzyme caspase through extracellular signaling;
- Endogenous pathway – activation of caspase by mitochondrial release of apoptotic enzyme activators.
These activated caspase degrade important proteins within the cell, causing apoptosis.
(i) Exogenous pathways
Activation of caspase (caspase):Caspase belongs to cysteine proteases, equivalent to ced-3 in nematodes, these proteases are the key enzymes that cause apoptosis, once activated by the signaling pathway, they can degrade the proteins in the cell, so that the cell is irreversibly dead. They all have the following features:
- Enzyme activity is dependent on the nucleophilicity of cysteine residues;
- The substrate is always cut off after aspartic acid (Asp, D), so it is named caspase (cysteine aspartate-specific protease), which is also called an apoptotic enzyme for convenience;
- Both are heterotetramers composed of two large and two small subunits, the large and small subunits are encoded by the same gene, and the precursors are cleaved to produce two active subunits.
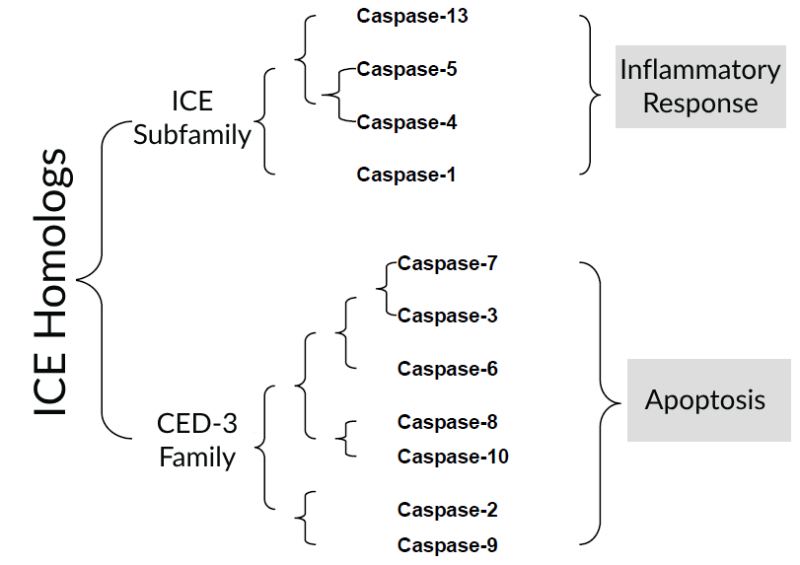
The earliest gene found to be homologous to Ced-3 in C. elegans in humans is the ICE (Interleukin-1β-converting enzyme) gene, which is named because of its ability to cleave interleukin precursors into active molecules. Through cDNA hybridization and search of genomic databases, 11 ICE homologs have been found in human cells, which can be divided into 2 subfamilies according to the number of homologies:
- ICE subfamily, including Capsase-1, Caspase-4, Caspase-5, and Caspase-13. Involved in inflammatory responses.
- CED-3 family, Capsase-2, Caspase-3, Caspase-6, Caspase-7, Caspase-8, Caspase-9 and Caspase-10. Involved in apoptosis. This family is divided into two categories according to their position and function in the apoptosis pathway: one is executioners or effectors, such as caspase-3, 6, and 7, which can directly degrade intracellular structural and functional proteins and cause apoptosis, but cannot be activated by autocatalytic or self-splicing; The other type is initiators, such as caspase-8 and 9, which can be activated by self-splicing after receiving a signal, and then cause a cascade reaction, such as caspase-8 can activate caspase-3, 6, and 7 in turn.
(ii) Endogenous pathways
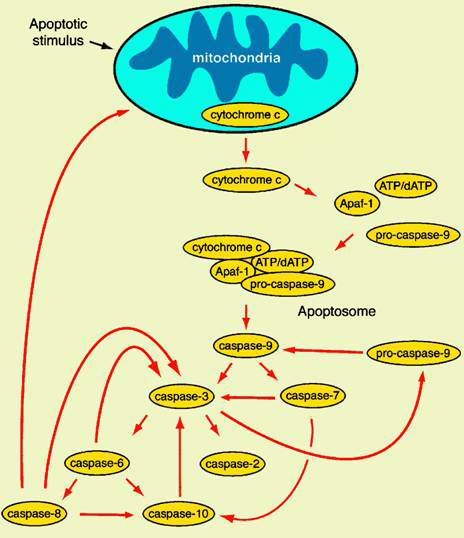
Mitochondria-mediated apoptosis: Cellular stress response or apoptosis signal can cause the release of mitochondrial cytochrome c, as an apoptosis-inducing factor, cytochrome c can form apoptosomes with Apaf-1, caspase-9 precursors, and ATP/dATP, and then convene and activate caspase-3, which in turn triggers the caspases cascade and leads to apoptosis.
It is generally accepted that cytochromes are released into the cytoplasm through mitochondrial PT pores (permeability transition pore) or mitochondrial transmembrane channels formed by members of the Bcl-2 family. Mitochondrial PT pore is mainly composed of proteins such as adenine nucleotide translocator (ANT) located in the inner membrane and voltage-dependent anion channel (VDAC) located in the outer membrane. Bcl-2 family proteins play a key regulatory role in the opening and closing of PT pores, pro-apoptotic proteins such as Bax can mediate the opening of PT pores through binding to ANT or VDAC, while anti-apoptotic proteins such as Bcl-2 and Bcl-xL can exert their anti-apoptotic effects by competitively binding to ANT with Bax, or directly preventing the binding of Bax to ANT and VDAC.
The structure of the Bcl-2 family is very similar to some toxins that form ion channels, such as E. coli toxins. Inserts into the membrane structure form larger channels that allow the passage of proteins such as cytochrome C, which may be another pathway for cytochrome C release.
Deletion mutations in Ced-3 and Ced-4 in C. elegans inhibit cell death at all developmental stages. In mammals, although mice with deletion of the Apaf-1 gene did not have caspase activation, most organ development was normal except for excess nerve cells. Recent studies have found that the proteins released with cytochrome c include Smac (second mitochondria-derived activator of caspase), apoptosis inducing factor (AIF) and endonuclease G (Endo G). Smac can bind to the BIR domain of IAPs (apoptosis inhibitory protein) through several amino acids at the N-terminus, thereby relieving the inhibition of caspase by IAP; AIF causes nuclear consolidation and chromatin breakage; EndoG can fragment DNA. It can be seen that even in the absence of caspase, apoptosis can still be caused by the mitochondrial pathway.
Among the cells that respond to Fas, type I cells, such as thymocytes, have sufficient activity of caspase-8 to cause apoptosis after being activated by Fas, and high expression of Bcl-2 in such cells cannot inhibit Fas-induced apoptosis. In type II cells, such as hepatocytes, Fas-mediated caspase-8 activation does not reach sufficient levels, so the apoptotic signal in these cells needs to be amplified by the apoptotic mitochondrial pathway. Activated caspase-8 cleaves Bid in the cytosol to form the active molecule tBid (truncated Bid), which enters the mitochondria, resulting in the release of cytochrome c, which amplifies the apoptosis signal.